Pulmonary hypertension (PH) is an important cause of morbidity and mortality in pediatric patients (1,2). PH is defined as an elevation of pulmonary artery pressure (PAP) due to any cause. Pulmonary arterial hypertension (PAH) is defined as a specific type of PH caused by pulmonary vascular disease of the precapillary arterioles in the absence of other causes
Diagnosis And Classification
PH is no longer classified as primary or secondary.
Currently, diseases are grouped according to similar pathology, pathophysiology, and treatment. A revision of the World Health Organization (WHO) classification, including most of the forms of PH encountered in children, was proposed at the WHO Dana Point meeting in 2008 however, this classification was not designed specifically for children. Group 1 is defined as PAH. This form of PH is due to pulmonary vascular disease of the precapillary arterioles in the absence of other causes (such as left heart disease—Group 2).
PAH is defined as a mean PAP > 25 mm Hg at rest, with a normal pulmonary capillary wedge pressure (<15 mm Hg) and increased pulmonary vascular resistance (PVR) index (>3 Wood units ! m)
Updated WHO Clinical Classification of Pulmonary Hypertension (Dana Point, 2008)
Pulmonary Arterial Hypertension
1.1 Idiopathic
1.2 Heritable
1.2.1 BMPR2
1.2.2 ALK1, endoglin (with or without hereditary hemorrhagic telangiectasia)
1.2.3 Unknown
1.4.1 Connective tissue diseases
1.4.2 HIV infection
1.4.3 Portal hypertension
1.4.4 Congenital heart disease
1.4.5 Schistosomiasis
1.4.6 Chronic hemolytic anemia
1.5 Persistent PH of the newborn
1 PVOD and/or PCH
2 PH due to left heart disease
2.1 Systolic dysfunction
2.2 Diastolic dysfunction
2.3 Valvular disease
3 PH due to lung diseases and/or hypoxemia
3.1 Chronic obstructive pulmonary disease
3.2 Interstitial lung disease
3.3 Other pulmonary diseases with mixed restrictive and obstructive pattern
3.4 Sleep-disordered breathing
3.5 Alveolar hypoventilation disorders
3.6 Chronic exposure to high altitudes
3.7 Developmental abnormalities
4 Chronic thromboembolic PH
5 PH with unclear and/or multifactorial mechanisms
5.1 Hematologic disorders: myeloproliferative disorders , splenectomy
5.2 Systemic disorders: sarcoidosis, pulmonary Langerhans cell histiocytosis, lymphangioleiomyomatosis, neurofibromatosis, vasculitis
5.3 Metabolic disorders: glycogen storage disease, Gaucher disease, thyroid disorders
5.4 Others: tumoral obstruction, fibrosing mediastinitis, chronic renal failure on dialysis
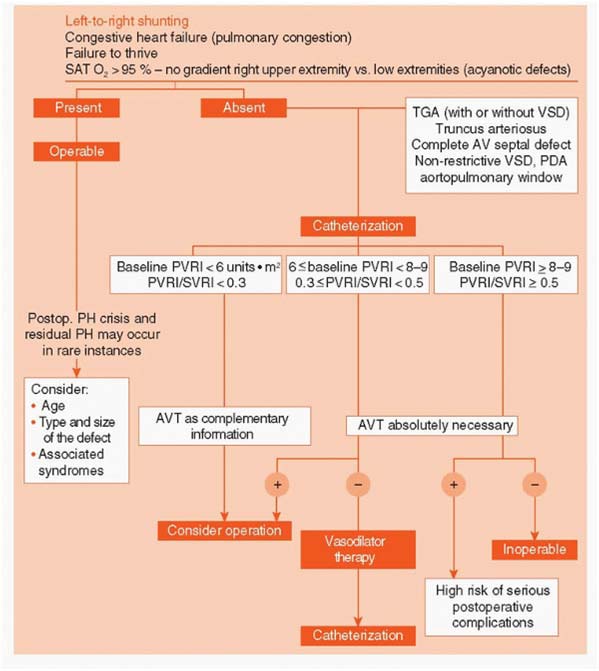
Clinical Classification of Congenital Systemic-to-Pulmonary Shunts associated with Pulmonary arterial Hypertension
Eisenmenger syndrome
Includes all systemic-to-pulmonary shunts due to large defects, leading to a severe increase of PVR and resulting in a reversed (pulmonary-to-systemic) or bidirectional shunt. Cyanosis, erythrocytosis, and multiple organs involvement are present.
PAH associated with systemic-to-pulmonary shunts
In these patients with moderate-to-large septal defects the increase of PVR is mild to moderate, systemic-to-pulmonary shunt is still largely prevalent and no cyanosis is present at rest.
PAH with small septal defects
In these cases with small defects (usually VSDs < 1 cm and ASD < 2 cm of effective diameter assessed by echo) the clinical picture is very similar to idiopathic PAH.
PAH after corrective cardiac surgery
Pulmonary Vascular Research Institute Classification of Pediatric Pulmonary Hypertensive Vascular Disease (Panama 2011)
- Prenatal or developmental pulmonary vascular disease
- Perinatal pulmonary vascular maladaptation
- Pediatric cardiovascular disease
- Bronchopulmonary dysplasia
- Pediatric lung diseases
- Multifactorial pulmonary vascular disease in congenital malformation syndromes
- Isolated pediatric PAH
- Pediatric thromboembolic disease
- Pediatric hypobaric hypoxic exposure
- Pulmonary vascular disease associated with other systemic disorder
Cardiac Lesions associated with Pulmonary Hypertension
1) Left-to-right shunts
Ventricular septal defect
Atrioventricular septal (canal) defect
Patent ductus arteriosus
Atrial septal defect
Aortopulmonary window
2) Increased pulmonary venous pressure
Cardiomyopathy
Coarctation of aorta
Hypoplastic left heart
syndrome
Shone complex
Mitral stenosis
Supravalvar mitral ring
Cor triatriatum
Total anomalous pulmonary venous return
Left ventricular outflow tract
Obstruction
Single ventricle
Norwood/Damus Stansel Kaye
Cavopulmonary anastomosis (Glenn)
Fontan procedure
Cyanotic heart disease
Transposition of the great arteries
Truncus arteriosus
Tetralogy of Fallot (pulmonary atresia/VSD)
Univentricular heart (high flow with/without restrictive atrial septum)
Anomalies of the pulmonary artery or pulmonary vein
Origin of a pulmonary artery from the aorta
Isolated pulmonary artery of ductal origin-unilateral"absence" of a pulmonary
artery
Scimitar syndrome
Palliative shunting operations
Waterston anastamosis
Potts anastamosis
Blalock-Taussig anastamosis
| Characterstic | Adults | Children |
|---|---|---|
| Main aetiologies | Idiopathic and familial PAH, thromboembolic and autoimmune diseases | Most common aetiologies outside immediate neonatal period are chronic lung disease and congenital heart disease |
| Histopathology | Advanced pulmonary vascular obstructive disease with intimal fibrosis and plexiform lesions | Severe pulmonary arterial medial hypertrophy with marked intimal proliferation |
| Presenting symptoms | Exertional dyspnoea, chest pain | Failure to thrivem poor appetite, cyanotic events, syncope, epilepsy-like attacks |
| The positive response rate to acture vasodilator testing with orally administered vasodilator drugs | About 12% | About 40% |
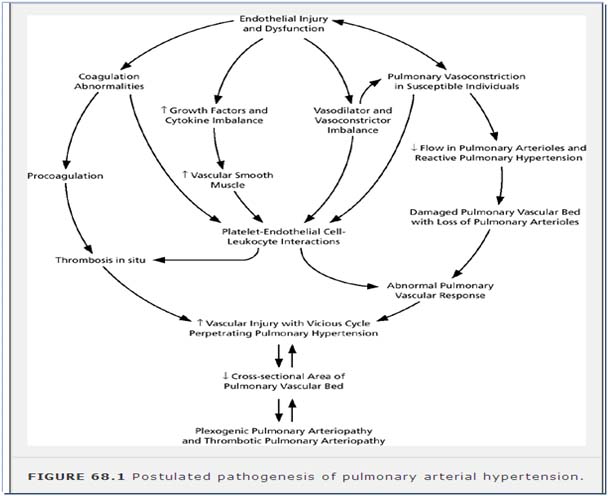
Genetic Predisposition
- Activation of the TGF--BMPR2 axis leads to suppression of proliferation and activation of apoptosis; conversely, these loss-of-function mutations exaggerate the susceptibility of vascular cells to proliferate.
- Mutations are usually in BMPR2 (bone morphogenetic protein receptor 2), and in ALK-1 (activin like kinase-1: receptor involved in hereditary hemorrhagic telangiectasia.)
Triggers for PAH
- 5-HT has been implicated in the pathogenesis of PAH by vasoconstriction and a mitogenic effect.
- Hypoxia inhibits voltage-gated potassium channels (Kv) in the PASMCs, opening voltage-gated calcium channels raising cytosolic Ca2, and initiating constriction.
Endothelin, an endogenous peptide, is a powerful pulmonary vasoconstrictor with mitogenic and fibrogenic effects. The vasoconstrictor effects are mediated by Ca2 channel activation and influx, and the growth and repair signals are transduced by activation of MAP kinases
Signs and Symptoms
The physical signs of PAH include a single and loud P2 without respiratory variation, right ventricular lift, and hepatomegaly with peripheral edema in the case of heart failure. In cases of severe disease, the P2 may be palpable. A diastolic murmur of pulmonary insufficiency, a holosystolic murmur of tricuspid regurgitation, or gallop rhythm may be audible.
In infants, symptoms are less specific and may involve poor appetite, failure to thrive,lethargy, diaphoresis, tachypnea, tachycardia, and irritability
Diagnosis
- ECG : RAE, RVH with strain , RAD
- CXR: peripheral pruning , RAE, RVH, calcification of central pulmonary arteries
- Echo: underlying cardiac defect, direction of shunt, abnormal motion of IVS (D shape LV )
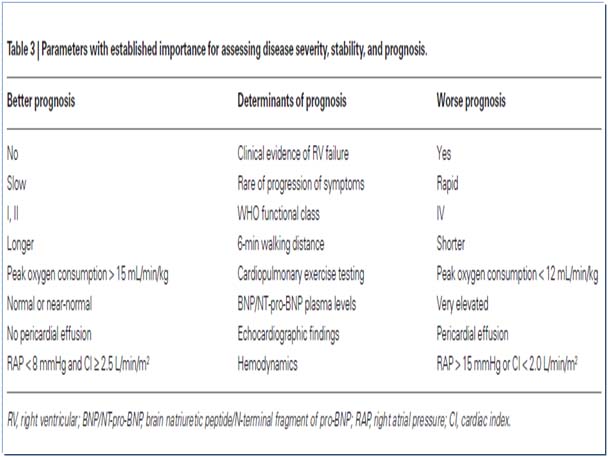
- Cardiac Catheterisation : pulmonary artery pressures, vasoreactivity of pulmonary vasculature by O2, NO, prostanoid. Acute response to vasoreactivity of pulmonary vasculature is defined as decrease in Mean PAP of at least 10mmHg with the mean PAP decreasing to or below 40mmHg with normal or high CO.
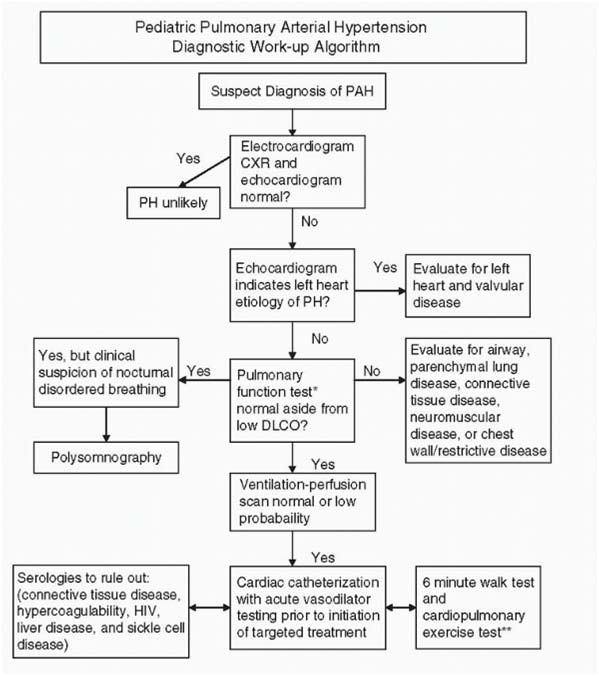
Evaluation – Algorithm Of Pah
Lab Analyses for Pulmonary Arterial Hypertension Workup
CBC,
Urinalysis,
Chem-20 including liver function profile, BNP/NTproBNP
Coagulation studies :
Factor VIII
Lupus anticoagulant
Protein C
Protein S
Factor II, V, VII
Factor V Leiden
Beta-2 Glycoprotein Antibodies
Cardiolipin IgG, IgM Ab
Antithrombin III
Prothrombin mutation
Platelet function assay
HIV test
Hepatitis profile
Thyroid function tests
CTD workup:
Lupus anticoagulant
ESR
ANA
Anti-DNA
Anticardiolipin antibodies
CH50 complement and components
Special ANAs
Anticentromere
Rheumatoid factor
Consider BMPR2 genetic testing
An algorithm of preoperative evaluation and management of pediatric patients with congenital cardiac shunts.
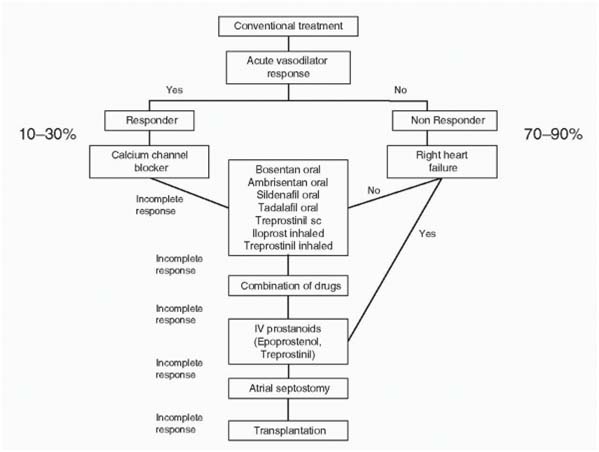
Treatment algorithm in children with severe PAH.
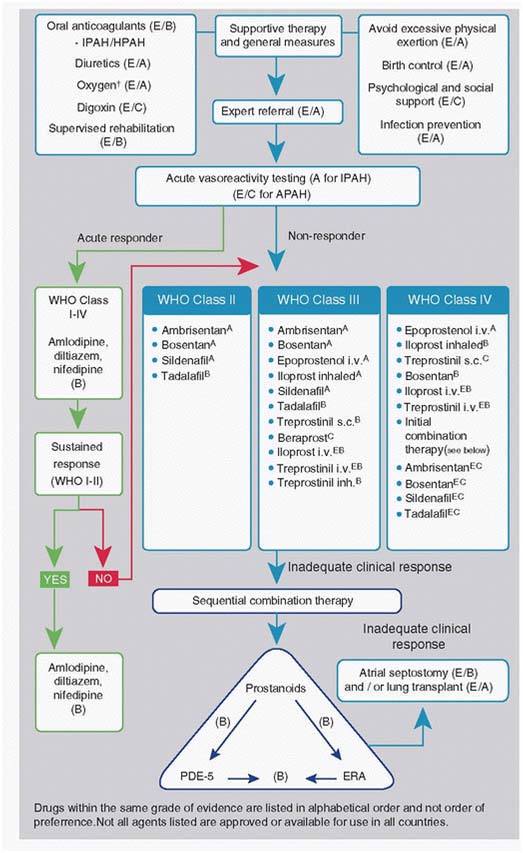
Treatment algorithm in adults with severe PAH
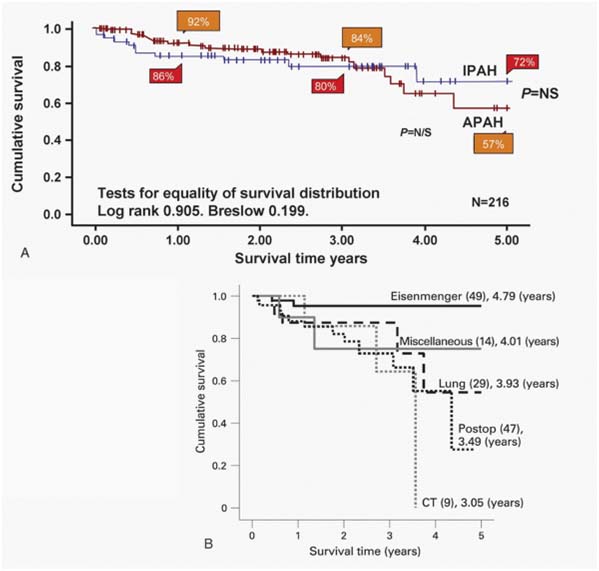
Survival curves for idiopathic pulmonary arteria hypertension (IPAH) and associated pulmonary arterial hyperten sion (APAH) cases censored for time in the study and for trans plantation. There was no significant difference between the two groups. B: Survival curves for the subgroups within the APAH group. The number in each group (brackets) and the predicted survival out of a possible 5 years are shown.
Treatment
- No cure for disease
- Search for Ideal treatment
- Rapid advancement in last decade
- Improved outcome
- A recent meta-analysis of 23 randomized controlled trials showed a 43% reduction in mortality and a 61% decrease in rate of hospitalizations with specific therapy.
- Still some patients have poor prognosis and rapid deterioration of the condition.
Goals of therapy are to improve
- quality of life
- functional class
- exercise capacity
- pulmonary hemodynamic, and
- long term survival for children with PAH.
General Management
- Avoid competitive sports
- Avoid isometric exercise
- Dehydration and excessive diuretics should be avoided
- Treat iron deficiency anaemia as there is association between IDA and TIA and stroke.
- Phlebotomy for symptomatic erythrocytosis
- Counsel against pregnancy; Pregnancy carries significant risk with Eisenmenger syndrome with maternal death reported up to 50%.
- Oral contraceptives should be avoided, surgical sterilization.
Medical Management
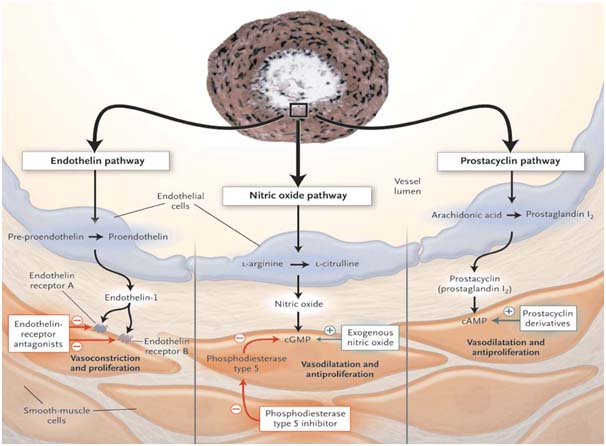
Three major pathways involved in abnormal proliferation and contraction of smooth muscle cells of pulmonary vasculature . These pathways are modulated by specific drugs.
- Prostacyclin pathway (Prostaglandin analogues)
- Endothelin pathway (Endothelin receptor antagonist)
- Nitric oxide pathway (PDE-5 inhibitor, and exogenous NO)
Sildenafil
- The use of sildenafil in infants and children with PAH is indicated in iPAH, PPHN refractory to treatment with iNO, to attenuate rebound effects after discontinuing iNO, and secondary PAH following cardiac surgery.
- Initial dose of 0.25-0.5 mg/kg every 4-8 hours is recommended for infants and children. Dose titration if needed and if tolerated to a dose of 1 mg/kg every 4-8 hours has been reported. Doses up to 2 mg/kg every 4 hours have been used in severe cases
Dosage of Bosentan in children
- <10 kg: 15.625 mg daily for 4 weeks, which is then increased to a maintenance dose of 15.625 mg twice daily;
- 10-20 kg: 31.25 mg daily for 4 weeks, then increased to a maintenance dose of 31.25 mg twice daily;
- 20-40 kg: 31.25 mg twice daily for 4 weeks, then increased to a maintenance dose of 62.5 mg BD;
- >40 kg: 62.5 mg twice daily for 4 weeks, then increased to a maintenance dose of 125 mg twice daily
Safety and tolerability
- Headache, dizziness
- Edema feet
- Increase in liver enzymes (ALT, AST): 5-8% of patients; monthly monitoring needed
- Pregnancy: Can cause birth defects, hence contraindicated. Not recommended during breast feeding
- Hemoglobin: levels can go down
Summary
- PAH in pediatrics, like in adults is a serious, progressive fatal condition.
- They differ in the underlying pathology and presence of age-specific conditions.
- PAH specific therapies have demonstrated clinical benefits and are safe and well tolerated in short term basis
- Early initiation and combination therapy may improve outcomes further in pediatric patients.
- Still waiting for ideal agent?!!!!!!!